Hiperconjugação
O termo hiperconjugação, também conhecido como efeito Baker-Nathan, foi utilizado pela primeira vez por John W. Baker e W. S. Nathan em 1935.[1][2][3]. Estes pesquisadores utilizaram o termo para explicar a lei da velocidade de reação em uma série de reações entre piridina e brometo de benzila substituído com grupos alquila na posição 4. Como os resultados não podiam ser explicados pelo efeito de campo, o novo conceito foi concebido e, a partir de 1940, com Robert S. Mulliken passou a ser amplamente utilizado.
No formalismo em que as ligações covalentes são divididas em σ (C-H, C-C, etc.) e π, hiperconjugação é a interação entre as ligações σ com uma rede de ligações π.[4]
Conceitos Gerais

Hiperconjugação é uma das maneiras que existem para descrever a deslocalização eletrônica nas moléculas. A mais conhecida e que tem grande importância na química orgânica ao longo de muitos anos é a conjugação. A conjugação se refere à deslocalização de elétrons Π em sistemas conjugados. Enquanto o termo hiperconjugação se refere à deslocalização de ligações sigmas.

A hiperconjugação pode ser uma resposta a estudos teóricos e experimentais que evidenciam significativas mudanças na geometria, na densidade de elétrons, em espectros de infravermelho (IR), e propriedades em ressonância magnética nuclear (RMN), podendo ainda influênciar em equilíbrios conformacionais, determinando a seletividade e as energias do orbital molecular (MO).

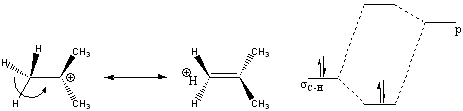
Segundo a teoria dos orbitais moleculares (MO), hiperconjugação é descrita como a interação entre orbitais eletrônicos, onde um deles corresponde a ligação sigma. O fenômeno é praticamente o mesmo que a conjugação, porém a interação tem que estar estabilizando um sistema, onde o orbital de mais alta energia tem que estar vazio ou parcialmente vazio (1 elétron), enquanto o outro de menor energia tem que estar parcialmente preenchido (Figuras 2 e 3).[5]
Analisando pela teoria da ligação de valência (VB), hipercojugação é a presença de estruturas de ressonância adicionais, como pode ser exemplificado pelo cátion terc-butila que apresenta maior estabilidade, devido à interação das ligações sigmas C-H com o centro deficiente de eletrons, do que metilas não substituídas.[6]

Tipos de Hiperconjugação
A hiperconjugação pode ser classificada de diferentes formas dependendo do número relativo de elétrons que contribui para a estabilização da ligação. Mulliken foi quem iniciou com estas classificações, e se referiu a hiperconjugação de duas maneiras: sacrificial (ordinária ou heterovalente) para sistemas neutros e isovalente (ou forte) para sistemas catiônicos, tendo ainda a possibilidade de ser para sistemas anionicos.


A Hiperconjugação positiva domina quando sistemas com orbitais p ou sistemas π fortemente aceptores ou sistemas sigmas fortemente doadores estão presentes na molécula. Isto é comum em cátions ou também moléculas neutras como boranas.
Um exemplo de efeito estabilizador por hiperconjugação positiva pode ser vista em estudos de β-sililetil cátions, que apresentam estabilidade em torno de 38 Kcal/mol em fase gasosa.[6]
Já a Hiperconjugação Negativa foi contactada em ligações C-F ao invés de ligações C-H, onde a polaridade se inverte.[7]

Basicamente, não se trata de doação de pares de elétrons de orbitais Π cheios (sendo isovalentes) ou podendo ser ainda pares solitarios de orbitais σ* (sendo sacrificial). Esta questão da hiperconjugação negativa foi explorada pelo cálculo dos índices de deslocalização eletrônica em tetrafluoretos de carbono.[8]
A hiperconjugação neutra se caracteriza por não ter efeito dominante entre aceptores e doadores de elétrons, tendo geralmente interações balanceadas. Embora ainda exista muitas controvérsias sobre este tipo de hiperconjugação, existem estudos que sugerem este tipo de estabilização em moléculas neutras ao analisar suas geometrias por raio-X.[9]
Extensão da deslocalização eletrônica por hiperconjugação
Assim como a conjugação, que é vista normalmente como formas de “comunicação” a distância entre orbitais que estejam conjugados, ou seja, como “pontes” para transportar elétrons dentro da molécula, a hiperconjugação, embora menos esclarecida, apresenta alguns relatos de deslocalização estendida[10][11], denominadas de homohiperconjugação e dupla hiperconjugação.
A homohiperconjugação é vista quando o centro insaturado entre aceptores e doadores de elétrons se invertem, ou quando o centro aceptor é um orbital p catiônico mais afastado do centro doador, ao se comparar com a cojugação. Já a dupla hiperconjugação amplia mais a deslocalização eletrônica quando existe uma ponte sigma entre o doador e o aceptor, sendo geralmente um orbital catiônico.[5]

Evidência e Discussão dos Efeitos
Lambert e Singer realizaram uma série de experimentos baseando-se nas constantes de acoplamento de ligação C-C para explicar a hiperconjugação sacrificial. Tal experimento consistia na obtenção de uma série de substâncias variando grupos doadores (M) e retiradores (X).

Onde, para X = H, Me e MeO, substratos foram preparados para todos os átomos M. Para X = CN, apenas M = Si e Sn foram preparados, e para X = NO2 apenas M = C foi preparado.
Através desse experimento eles observaram, como esperado, que a hiperconjugação aumenta a ordem de ligação entre os carbonos benzílicos e ipso (CH2-Ci) e diminui a ordem de ligação entre o átomo doador e o carbono benzílicos (CH2-M) e em termos de estrutura, o comprimento de ligação CH2-Ci diminui e aumenta a constante de acoplamento e para comprimento de ligação CH2-M ocorre o inverso. Deve-se contudo salientar que as constantes de acoplamento C-C dentro do anel aromático são essencialmente independentes das mudanças no M. Dessa forma, os fatos experimentais vão contra a hipótese original de Baker-Natan, visto que a hiperconjugação não resulta em alternância de títulos de comprimento dentro do anel aromático.
A hiperconjugação tem aparecido na literatura podendo ser utilizada direta ou indiretamente como um parâmetro na correlação de reatividade química ou propriedade física, sendo assim desde a correlação proposta por Kreevoy e Taft, adaptações dessa correlação tem sido feita para que os efeitos de parâmetros estéricos possam ser incluidos.[12]
Schubert e Sweeney relataram efeitos físicos e químicos para comentar sobre hiperconjugação, porém observaram a ocorrência de contradição entre evidências químicas e físicas de modo que passaram a levar em conta o solvente. Assim, chegaram a conclusão de que não é que a hiperconjucação não tenha consequências, mas se o papel do solvente é importante, então outros modos de liberação de elétrons podem ser relativamente mais importantes.[13]

Siehl et. al. Investigou a importância da conjugação e da hiperconjugação na estabilização de cátions 1-(p-anisil)vinil beta substituídos, usando espectroscopia de RMN e cálculos computacionais.
No cátion substituído 1-(p-anisil) existe uma barreira torsional para a rotação do grupo metóxi em torno da ligação fenil-C4-oxigênio com a deslocalização da carga positiva no grupo metóxi, resultando numa ligação dupla.

Os cálculos computacionais mostraram que a ligação O-Me é coplanar com o anel arila como resultado da contribuição das formas de ressonância. Quando o substituinte passa de beta-H para beta-Si há a diminuição das distâncias de ligação C4-OMe e o aumento dos ângulos de ligação C4-O-Me demonstrando que a dispersão da carga para a grupo p-metoxi tornou-se mais importante como interação por hiperconjugação.
Segundo Siehl et. al. o grupo O-Me é orientado perpendicularmente ao plano do anel e não sofre conjugação. Os cálculos confirmam que a doação de elétrons por hiperconjugação segue a ordem: C-Si> C-C> C-H.
A combinação de métodos teóricos e experimentais usados no estudo revelou que o deslocamento químico para C4 e a barreira de rotação para C4-O determinados experimentalmente e através de cálculos, assim como parâmetros geométricos foram úteis para investigar a estabilização por ressonância do sistema pi e a estabilização por hiperconjugação das ligações beta-sigma dos tipos de carbocátions.[14]
Kreevoy e Taft[15] sugeriram que a hiperconjugação foi um fator importante na reatividade química e que um parêmetro que caracteriza a hiperconjugação poderia ser utilizado com outros parâmetros para correlacionar os valores de log k, mas uma reanálise desses dados mostraram que em todos os casos estudados bons resultados poderiam ser obtidos apenas considerendo-se os efeitos elétricos, estérico e os efeitos de polarizabilidade, não havendo necessidade de envolver a hiperconjugação a fim de explicar satisfatoriamente os dados obtidos, pelo contrário o melhor seria abandonar o uso dos parâmetros de hiperconjugação, considerado desnecessário por muitos autores.
A hiperconjugação tem aparecido na literatura podendo ser utilizada direta ou indiretamente como um parâmetro na correlação de reatividade química ou propriedade física, sendo assim desde a correlação proposta por Kreevoy e Taft, adaptações dessa correlação tem sido feita para que os efeitos de parâmetros estéricos possam ser incluidos.[16]
Em 2006, Ingold e DiLabio propõem que a correlação entre as energias de dissociação (BDE’s) de C-H e os termos de deslocalização de spin calculados a partir do espectro de EPR evidenciam que a hiperconjugação tem um papel fundamental na estabilização dos radicais alquilas. Segundo o relato o decréscimo das entalpias de ligações (BDE) C-H ao longo da série H3C-H > H3C-CH2-H > (H3C)2CH -H > (H3C)3C –H tem sido convencionalmente atribuído ao efeito doador de grupos β-H3C para o centro radical via hiperconjugação. Ainda, segundo o autor, a hiperconjugação no radical é um fato atestado pela espectroscopia de ressonância paramagnética eletrônica. Por exemplo, o radical etila tem significativa densidade de spin desemparelhado nos três hidrogênios do grupo β-H3C. Na medida em que este spin aumenta por deslocalização hiperconjugativa do elétron desemparelhado, ele estabiliza mais o H3-CCH2* do que o H3C*. Assim, usando a constante de acoplamento hiperfino obtido a partir do EPR (vide tabela) ele demonstra que a hiperconjugação contribui para o decréscimo do BDE da ligação C-H, para a série metano, etano, propano e isobutano de maneira mais ou menos quantitativa, conforme mostrado no gráfico.[17]
Porém, Scott Gronert refuta a afirmação dada por Ingold e DiLabio. Segundo ele a correlação proposta não pode ser estendida para os BDE’s de C-C, conforme mostrado no gráfico, e a natureza do desvio é inconsistente com os atuais dados das energias de interação 1,3-carbono-carbono em alcanos. Assim, para o autor, os resultados sugerem que a hiperconjugação não é o fator chave na determinação dos BDE’s.[18]
Fatores que controlam a hiperconjugação
Overlap e simetria orbitalar
Para que se tenha uma boa interação, os orbitais vizinhos devem ser coplanares, e ainda, estudos tem demonstrado que o arranjo geométrico antiperiplanar é preferencial frente ao sinperiplanar. Este efeito é evidenciado, por exemplo, na maior estabilidade da conformação trans do butadieno frente a conformação cis.
As característi as conformacionais, estruturais e reacionais tem grande interferência do efeito de hiperconjugação. Um ponto importante das interações que envolvem hiperconjugação é a sua componete estereoeletrônica. Tais interações dependem da sobreposição de orbitais (formando, preferencialmente, ligações sigmas).
As forças que controlam a barreira de rotação em torno das ligaçãos são usadas comumente para a análise conformacional de uma dada estrutura. O etano é um dos sistemas mais utilizados em estudos dessa natureza. A menor energia da estrutura alternada frente a estrutura eclipsada do eteno (cerca de 3 kcal/mol para essa barreira de rotação), tem sido, geralmente, atribuído a estéreo repulsão entre os elétrons da ligação C-H [19]. Mulliken, já em 1939, supôs que a hiperconjugação desempenha um papel fundamental no potencial de rotação tanto do etano, como de outras moléculas [20].

Pophristic e Goodman [21], utilizaram análise NBO para estudar as contribuições dos três principais efeitos que contribuiem para a estabilização de uma coformação estrutural para o etano: intarações estéricas e de hiperconjugação. Eles descobriram que quando retiram as interações de hiperconjugação, a estrutura é melhor estabilizada em uma conformação preferencialmente eclipsada.
Para interações intermoleculares por hiperconjugação negativa, o melhor arranjo estereoeletrônico envolve um arranjo colinear onde o orbital doador interage com a parte posterior do lóbulo sigma* [22], essa característica reflete reações do tipo SN2 (transferência de elétrons).

Para hiperconjugação positiva, a geometria possível não se restringe ao lobo posterior da ligação sigma. A coordenação de um eletrófilo leva a um ataque frontal e um processo de clivagem por SE2.

Efeitos Estereoeletrônicos
Barreira rotacional: Etano
A busca para determinar e interpretar a barreira rotacional interna das moléculas foi inicialmente estudado por Pitzer e Lipscomb (LP) [23] Eles estudaram a molécula do etano, nas conformações escalonadas(S) e eclipsada (E) (figura 18) considerando que o comprimento de ligação e ângulos de ligação nessas conformações são iguais.[24][25]

O baixo valor para a barreira rotacional do etano, 3,3 kcal/mol [23] foi atribuído ao efeito estérico de repulsão entre os elétrons na ligação C-H na conformação de eclipse. A rotação, que causa o enfraquecimento da ligação C-C e a hiperconjugação, são os fatores responsáveis para a alta estabilidade na conformação escalonada.[26][26][27][28] Segundo Pophristic e Goodman usando análise Natural bond orbitals (NBO), encontraram que a conformação em forma de eclipse é a estrutura majoritária.[29]. Esse estudo entretanto foi contestado recentemente [30], mostrando que há um artefato nos cálculos NBO.
Através da análise de NBO é possível analisar as interações hiperconjugativas com mais detalhes do que quando analisado por Molecular orbital (MO).[31] O modo de hiperconjugação para a conformação escalonada é atribuído à estéreo-eletrônica anti-periplanar.(figura 19)
Figura 19.
Hiperconjugação neutra: Alcenos e alcinos
Existem controvérsias a respeito da hiperconjugação neutra, porém alguns trabalhos baseados em dados termodinâmicos ressaltam a importância da hiperconjugação em alcenos e alcinos. Roger et al[32][33] observou que a estabilidade atribuída a conjugação π do composto 1,3 butadiino é praticamente zero quando estimado através da abordagem clássica de Kistiakowsky et al[34]. Kistiakowsky et al sugeriu que a estabilidade do 1,3 butadieno pode ser avaliada através da liberação energética da reação de hidrogenação do composto como mostra o esquema ao lado:

De acordo com Kistiakowsky et al a reação de hidrogenação do 1,3 butadieno a 1-buteno é menos energética que a reação de hidrogenação do 1-buteno a butano. Kistiakowsky et al associa esse fato a maior força da conjugação π no composto 1,3 butadieno. Já para 1,3- Butadiino foi observado que a liberação energética nas duas etapas de hidrogenação (ver esquema) foi igualmente exotérmica.
Segundo Kistiakowsky et al esses resultados sugerem que a conjugação do 1,3 Butadiino não interfere na estabilidade do composto e ressalta a importância da hiperconjugação neutra para explicar tal fato. Ao analisar o efeito da hiperconjugação através do método citado podemos observar que o efeito no 1.3 butadiino é mais forte em relação ao 1,3 butadieno.Jarowski et al [35] também atribuiu a estabilidade do 1-butino e 1-buteno a hiperconjugação neutra da molécula.
Frenking e coworver[36] com base em avaliações de deslocalização π em alcenos e alcinos relatou, que a força de estabilidade relacionado a hiperconjugação é duas vezes maior que a força relacionada a conjugação π.
Equilíbrio conformacional
Pode-se citar ainda como exemplo da importância da hiperconjugação neutra em propeno (pode ser fornecido por seu perfil conformacional). Para o propeno a conformação mais estável é o “eclipsado”, pois uma ligação metil C-H eclipsa a ligação com o carbono vizinho σC-C. A conformação escalonada em uma ligação metil C-H eclipsa os carbonos vinil adjacentes da ligação C-H, é menos estável por aproximadamente 2 kcal/mol,[37]. Estas nomenclaturas são inadequadas pois a conformação“eclipsada” do propeno é estereoeletronicamente semelhante à conformação escalonada do etano (figura 18) e vice-versa.
Lin et al.[38] comprovaram por análises NBO (análises dos orbitais naturais de ligações) que a hiperconjugação é a mais importante interação vista entre os grupos metil e vinil e divide-se em três partes: a interação C=C πCH3 → π*, a interação C=C π* CH3 → πC e as interações entre orbitais vizinhos no plano σC-H do grupo metil e σ*- orbital do vinil antiperiplanar da ligação C-H. Um esclarecimento análogo foi apresentado por Basso et. al.[39], sobre a proveniência das preferências conformacionais em compostos carbonilados.
 Figura 21. Diferença entre as conformações eclipsada e escalonada do propeno.
Figura 21. Diferença entre as conformações eclipsada e escalonada do propeno.
Capacidade de acepção das ligações sigma: Os papéis opostos da eletronegatividade e da energia do orbital
A capacidade aceptora de ligações sigma C-X em etanos monossubstituídos com relação ao mesmo doador (ligação C-H antiperiplanar) aumenta em direção ao fim de um período e para baixo nas famílias. O aumento da capacidade aceptora de ligações sigma C-X em períodos, paralelamente ao aumento eletronegatividade, como resultado de mudanças favoráveis na polarização s*, aumenta a matriz Fock e elementos de sobreposição da matriz para a interação [65 e 129]. Por outro lado, essa tendência é oposta quando se pensa nas famílias, já que a capacidade aceptora aumenta para baixo, e a eletronegatividade para cima. Essas comportamento pode ser compreendido a partir da equação da análise NBO (Natural Bond Orbital). A ordem relativa de capacidade aceptora (a energia da interação sC-H -> s*C-X é dada entre parênteses) é a seguinte: X = Br (6,3) > Cl (6,2) > SH(1) (5,4) > F (5,1) > OH(1) (4,7) = SH(2) (4,7) = SeH (4,7) > PH2(1) (4,6) > AsH2 (4,5) = NH2(1) (4,5) > OH(2) (4,2) > PH2(2) (4,0) >NH2(2) (3,8) = GeH3 (3,8) > SiH3 (3,6) > CH3 (3,4) > H (3,2). Dois valores para alguns substituintes correspondem a diferentes conformações. Curiosamente, efeitos estereoeletrônicos exibidos por ligações C-X com X pertencente ao segundo e terceiro períodos são altamente anisotrópicas. Ligações C-calcogênio, por exemplo, são excelentes aceptores-s e na ponta referente ao carbono, mas péssimos aceptores-s no terminal do calcogênio.[40][41]
Propriedades doadoras de pares solitários
As diferenças relativamente sutis nas energias envolvidas na hiperconjugação tornam-se mais pronunciadas e quimicamente significativas em interações envolvendo melhores doadores, como o par solitário do nitrogênio. Os dados NBO de a-halogenoaminas indicam que tanto a alta energia do orbital não-ligante e sua alta polarizabilidade contribuem para uma energia de interação incrementada.
Hibridização em pares solitários
As diferenças em hibridização são particularmente importantes para interações estereoeltrônicas hiperconjugativas por várias razões. Primeiro, a hibridização é diretamente relacionada à geometria molecular, e determina os ângulos e direção nos quais os orbitais não-ligantes são projetados no espaço para a sobreposição com os orbitais aceptores. Segundo, ela controla o tamanho relativo dos dois lobos de um par solitário. Os lobos frontal e posterior são equivalentes para pares puramente p, enquanto o posterior diminui de tamanho com o decréscimo do caráter p no híbrido spn. Terceiro, a hibridização do orbital doador é relacionada a sua energia absoluta. Um incremento no caráter p leva a um aumento na energia do orbital que diminui a diferença energética entre o par solitário doador e um orbital aceptor s* ou p*. Em geral, a capacidade doadora é diretamente proporcional ao caráter p do par solitário. Pares solitários com 100% de caráter p são melhores doadores que doadores hibridizados spn.
Energia dos pares solitários
A energia de interação é inversamente proporcional à diferença de energia, que depende da energia relativa do par solitário. As tendências na energia de pares solitários podem ser prontamente compreendidas em termos de sua hibridização e da eletronegatividade de X. Aumento na eletronegatividade e diminuição no caráter p abaixam a energia orbital do par solitário. Embora o oxigênio seja mais eletronegativo que o nitrogênio, a hibridização puramente p axial deste, comparada à do nitrogênio, tem um efeito menor. Nesse caso, o efeito da hibridização compensa a diferença de eletronegatividade.
Efeito Anomérico
Histórico
Originalmente, define-se efeito anomérico como a tendência de heteroátomos substituintes, adjacentes a um heteroátomo formador de um ciclohexano saturado cíclico, em assumir uma orientação axial ao invés de uma orientação equatorial, menos impedida estericamente.[42] Esse termo foi utilizado na literatura porque envolve o Carbono 1 de derivados de piranoses e de glicopiranosil, chamados Carbonos Anoméricos.

O efeito anomérico é um dos fenômenos que é classicamente explicado pela hiperconjugação. Nesses casos a hiperconjugação ocorre pela interação do par de elétrons livres do heteroátomo do ciclo com o orbital antiligante da ligação entre o carbono anomérico e seu substituinte.[43] Com a ajuda da Química Computacional trabalhos recentes mostram que provavelmente a hiperconjugação tem pouca, ou nenhuma, contribuição para a existência desse efeito, sendo que a estabilização dos substituintes na posição axial é melhor explicada pelos efeitos das interações eletrostáticas e estéricas dos grupamentos da própria molécula.[43][44] Apesar disso encontra-se na literatura evidências de que a hiperconjugação pode explicar o efeito anomérico em compostos acíclicos, como carbenos, estabilizados na posição antiperiplanar.[45]
Modelos
Tradicionalmente, esse efeito é explicado tanto pelo conceito de estabilização do momento dipolar [46][47], como também pelo efeito de hiperconjugação, através da estabilização gerada pela interação do par de elétrons livres do heteroátomo com o orbital antiligante da ligação entre o carbono anomérico e seu substituinte [48]. Essas explicações, especialmente a que diz respeito ao efeito de hiperconjugação, são objetos de controvérsia em publicações recentes [43].
O modelo eletrostático mostra que a estabilidade do substituinte eletronegativo na posição axial ocorre devido à diminuição da desestabilização que ocorre quando o momento dipolar resultante entre o dipolo da ligações entre o C1 e o heteroátomo polar substituinte na posição axial e da ligação entre o heteroátomo do anel e o C1 não possui um caráter aditivo (estrutura A), como aconteceria se o substituinte estivesse na posição equatorial (estrutura B). Os dipolos alinhados causam um efeito aditivo desestabilizando a molécula e, portanto, aumentando a energia e, consequentemente, assumindo uma conformação menos estável.

Lemieux et al. [49] publicou em 1969 em seu artigo, um experimento que demonstrava que a conformação espacial do 2-metoxitetrahidropirano variava conforme a polaridade do solvente utilizado, de forma que solventes mais polares estabilizavam melhor grupos polares na posição equatoriais do que solventes não-polares (Figura 22). No entanto, esse comportamento não era verificado em certos sistemas.

Juaristi et al. [50] demonstrou em seu trabalho que para a molécula 2-carbometoxy-1-3-ditiano, sob baixas temperaturas, a conformação axial é mais favorável termodinamicamente. Assim, o efeito eletrostático não seria suficiente para explicar o efeito anomérico.
Segundo Wolfe e outros pesquisadores [51][52], a preferência na orientação axial ocorre em consequência de um efeito mais geral, que requer que um par de elétrons isolados nX de um heteroátomo X e a ligação C–Y em um sistema YCH2X estejam alinhados numa geometria antiperiplanar que maximize a interação de hiperconjugação n(X) → σ*C–Y.


Embora existam diversos componentes no efeito anomérico, como os componentes eletrostáticos (interações dipolo-dipolo e efeitos estéricos, por exemplo), as interações de hiperconjugação de orbitais antiperiplanares exercem um papel particularmente importante quando observamos (1) as mudanças estruturais (alongamento da ligação C–Y e diminuição da ligação C–X), (2) a densidade de distribuição eletrônica (aumento de carga negativa sobre Y) e (3) a reatividade (enfraquecimento da ligação C–Y). Uma interação semelhante entre o par isolado de um heteroátomo exocíclico Y e o orbital σ*C–X do anel oferece a base estereoeletrônica para o chamado Efeito Exo-anomérico.

Definição Moderna
Atualmente, o efeito é considerado um caso especial da preferência genérica (Efeito Anomérico Generalizado) de um substituinte no Carbono anomérico estar em posição sinclinal (gauche) em sistemas X–CH2–Y–CH2, onde X e Y são heteroátomos com pares de elétrons não-ligantes, usualmente sendo pelo menos um deles Nitrogênio, Oxigênio, Enxofre ou Flúor. Essa definição mais genérica do efeito anomérico permite a aplicação em moléculas acíclicas.

Radicais

Tetrahidrotiopiranos

Dioxanos e Ditianos

Nitrogênio de aminas

Centros de hibridização sp2

Efeito Anomérico Reverso

Segundo Cramer [53], cálculos em fase gasosa são consistentes com uma grande deslocalização por hiperconjugação da densidade do par de elétrons isolados do Oxigênio do anel de Tetrahidropirano no orbital axial da ligação C-N(+)σ*.
Efeito Homoanomérico

Os papéis relativos aos efeitos homoanoméricos W- e Plough em aza-, oxa-, tio e selenoheterociclos foram investigados em experimentos com RMN13C e análises NBO, e revelaram que tais efeitos desempenham um importante papel na tendência relativa na constante de acoplamento de uma ligação 1J(C-H), necessária no entendimento das propriedades conformacionais de carbohidratos, azacarbohidratos e outros substratos de interesse biológico [54].
Embora os efeitos homoanoméricos sejam considerados mais fracos que as clássicas interações vicinais anoméricas, sua importância aumenta significantemente quando a habilidade aceptora dos orbitais σ* aumentam em consequência do alongamento da ligação e/ou polarização. Por exemplo, a solvólise de piperidinas e pirrolidinas com um grupo de saída no Carbono β se dá através da formação cátions aziridínicos cíclicos, devido a ajuda anquimérica do par isolado do Nitrogênio [55].
Considerações Finais
Trabalhos recentes [42] afirmam que, através da utilização de modelos semi-empíricos, neste caso DFT (Density Functional Theory [56]), não há relação estatisticamente relevante entre hiperconjugação e efeito anomérico, sendo a diferença de energia entre os anômeros um efeito decorrente da diferença do momento dipolo. A decomposição em DFT e a nova análise de Densidade Funcional Estérica mostram que três componentes energéticos (correlação de mudança, interação eletrostática clássica e energia de densidade funcional estérica) são diretamente proporcionais à diferença total de energia entre os isômeros axial e equatorial, indicando que na posição equatorial o heteroátomo substituinte sofre uma grande energia de correlação de troca (exchage-correlation) e de interação eletrostática, porém baixo impedimento estérico. Entretanto, todas essas correlações não são fortes o suficiente para garantir uma explicação eloquente para o mecanismo geral do efeito anomérico. Segundo o autor, é o baixo impedimento estérico e forte interação eletrostática que governa a estabilidade conformacional.
Ao mesmo tempo vem aparecendo na literatura evidências de que a hiperconjugação pode explicar o efeito anomérico em compostos acíclicos, como carbenos, na posição antiperiplanar [57].
Ligações de Hidrogênio
Apesar das ligações hidrogênio serem um fenômeno complexo e de muitos fatores poderem estar envolvidos na formação de complexos do tipo X-H - - - Y conectados por ligações hidrogênio, a hiperconjugação negativa n(Y) → σ∗ X-H (a qual é comumente chamada ‘componente covalente’ ou ‘componente de transferência de carga’) é um dos dois maiores efeitos estabilizadores da ligação hidrogênio, juntamente com a interação eletrostática entre dipolos [58]. A importância de interações hiperconjugativas de um par isolado do aceptor de ligação hidrogênio com o orbital σ∗ X-H do doador é bem documentada por análise energética NBO [59]. Como tais interações levam a um aumento na população do orbital antiligante X-H, elas alongam a ligação X-H. Quando o componente hiperconjugativo da ligação hidrogênio é fraco, o efeito de alongamento da ligação citado acima pode ser compensado por repolarização e rehibridização da ligação e então acontece a formação das chamadas ligações hidrogênio “blue-shifting” [60].

Efeito Gauche
O efeito gauche ou “efeito gauche do fluoreto” é a preferência pela conformação Gauche (ou sinclinal) em etanos 1,2-dissubstituídos (do tipo X-C-C-Y) quando X e Y são dois aceptores [61][62].

Apesar de X e Y serem geralmente F e O, a escolha do segundo substituinte (Y) é bastante ampla em fluoroetanos (X = F) e estudos utilizando DFT mostram esse efeito [63]. O efeito gauche pode ter origem meramente baseada na interação hiperconjugativa e estereoeletrônica, bem como pode ser baseado em interações eletrostáticas dipolo-dipolo ou carga-dipolo.

O efeito gauche é útil para o controle da estrutura e reatividade química, como demonstrado por Schuler et al.[64] para peptídeos e por Tavasli et al. para ácidos graxos [65]. O papel dominante da doação hiperconjugativa antiperiplanar σC-H → σ∗ C-X e σC-H → σ∗ C-Y nesse efeito foi mostrado com análise NBO [66]. Para X = Cl, Br e I, a conformação anti é mais estável do que a gauche devido ao fato de que as ligações dos halogênios mais pesados são bons doadores, além de aceptores e as interações σC-Y → σ∗ C-X se tornam suficientemente grandes para competir com as interações σC-H → σ∗ C-X. A preferência pela conformação sinclinal em sistemas acíclicos do tipo X-C-Y-C também é comumente tratada como efeito gauche, mas é na verdade um exemplo de efeito anomérico dominado por interações n → σ∗. De maneira análoga, isômeros cis do 1,2-difluoreteno e 1,2-dicloroeteno são experimentalmente mais estáveis do que os isômeros trans (0.9 e 0.43 kcal/mol) enquanto os isômeros do 1,2-dibromoetano têm estabilidade igual dentro do erro experimental. Esse fenômeno, extensamente analisado é chamado de efeito ‘cis’ e é bastante similar ao efeito gauche mostrado [67][68]. Recentemente Yamamoto et al. demonstraram as contribuições de deslocalização eletrônica e repulsão estérica utilizando a teoria NBO no nível MP2/6–311++G(3df,3pd), capaz de reproduzir as diferenças experimentais de energia entre os isômeros geométricos [69]. Dois mecanismos de deslocalização, hiperconjugações periplanares (efeitos sinperiplanar e antiperiplanar) e deslocalizações do par isolado para os orbitais antiligantes da ligação C-C (efeito de deslocalização do par isolado) foram vistos como as forças de estabilização ‘cis’. Apesar da preferência estereoeletrônica comum para o arranjo anti do melhor doador e do melhor aceptor dominados por X = F, a diferença nas energias das interações antiperiplanares diminui para os isômeros dos halogênios mais pesados onde as interações σC-X → σ∗ C-X aumentam em importância relativa devido à maior capacidade de doação das ligações σC-Cl e σC-Br. Interessantemente, apesar das interações hiperconjugativas dos orbitais σ claramente favorecerem os isômeros cis, as interações dos pares isolados com os orbitais π e σ da ponte possuem um efeito ainda maior [60].
Referências
- ↑ BAKER, W. J.; NATHAN, W. S.; The mechanism of aromatic side-chain reactions with special reference to the polar efects of substituents. The effect of unipolar substituents on the critical energy and probability factors in the interaction of benzyl bromide with pyridine and α-Picolinc in various solvents. Journal of the Chemical Society, (1935), 519-27.
- ↑ 2. BAKER, W. J.; NATHAN, W. S. The mechanism of aromatic side-chain reactions with special reference to the polar efects of substituents. Part IV. The mechanism of quaternary salt formation. Journal of the Chemical Society, (1935), 1840-44.
- ↑ 3. BAKER, W. J.; NATHAN, W. S.; The mechanism of aromatic side-chain reactions with special reference to the polar efects of substituents. Part V. The polar efects of alkyl groups. Journal of the Chemical Society, (1935), 1844-47.
- ↑ IUPAC, Compêndio de Terminologia Química, 2ª ed. ("Gold Book"). Compilado por A. D. McNaught e A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Versão online: "hyperconjugation" (2006–) criado por M. Nic, J. Jirat, B. Kosata; atualizações compiladas por A. Jenkins. ISBN 0-9678550-9-8.
- ↑ a b ALABUGIN, I. V., GILMORE, K. M., PETERSON, P.W., Hyperconjugation, Advanced Review, Vol. 1, 2011.
- ↑ a b HAVENITH, R. W. A., VAN LENTHE, J. H., Delocalization in Valence Bond—Hyperconjugation. International Journal of Quantum Chemistry, Vol 109, 2426–2429 (2009).
- ↑ LAMBERT, J. B., Singer, R. A., Neutral Hyperconjugation. J. Am. Chem. SOC. 1992, 114, 10246-10248.
- ↑ ROBERTS, J. D., WEBB, R. L., McElhill, E. A. J. Am. Chem. Soc., 1950, 72, 408–411.
- ↑ WIBERG, K. B., RABLEN, P. R., Origin of the Stability of Carbon Tetrafluoride: Negative Hyperconjugation Reexamined. J. Am. Chem. Soc. 1993, 115, 614-625.
- ↑ LAUBE, T. Detection of hyperconjugative Effects in Experimentally Determined Structures of Neutral Molecules. J Am Chem Soc, 1988, 110:5511– 5517.
- ↑ SCHERPES, T., MICH, J. Optimized ladder C and ladder H models for sigma conjugation: chain segmentation in polysilanes. J. Phys. Org. Chem., 2002, 15:490–498.
- ↑ LAMBERT, J. B., SINGER, R. A., Neutral Hyperconjugation. J. Am. Chem. SOC. 1992, 114, 10246-10248.
- ↑ SCHUBERT, W. M.,SWEENEY, W. A., Concerning the Baker-Nathan Effect. Department of Chemistry ow the University of Washington,1955,21,119-124.
- ↑ SIEBL, H.,KAUFMANN, F.,HOD, K.,NMR Spectroscopic and Computational characterization of 1-(p-Anisy1)vinyl Cations. Methoxy Group Rotation as a Probe of Cβ-Si, Cβ-C, and Cβ-H Hyperconjugation.J. Am. Chem. SOC. 1992,114, 9343-9349.
- ↑ KREEVOY, M. M.; TAFT, R. W. J. Am. Chem. SOC. 1955, 77, 5590;1967,79,4011
- ↑ CHARTON,M.,CHARTON, B.I.,Hyperconjugation as a Parameter in Correlation Analysis.J. Org. Chem., Vol. 47, No. 1, 1982,8-13.
- ↑ Organic Letters de 2007 (DOI:10.102/ol062293s)
- ↑ Organic Letters de 2007 (DOI:10.1021/ol070706z)
- ↑ Sovers OJ, Kern CW, Pitzer RM, Karplus M. Bondfunction analysis of rotational barriers: ethane. J Chem Phys 1968, 49:2592–2599. Christiansen PA, Palke WE. A study of the ethane internal rotation barrier. Chem Phys Lett 1975, 31:462–466
- ↑ Mulliken RS. Intensities of electronic transition in molecular spectra. IV. Cyclic dienes and hyperconjugation. J Chem Phys 1939, 7:339–352
- ↑ Pophristic V, Goodman L. Hyperconjugation not steric repulsion leads to the staggered structure of ethane. Nature 2001, 411:565–568
- ↑ ALABUGIN, I. V., GILMORE, K. M., PETERSON, P.W., Hyperconjugation, Advanced Review, Vol. 1, 2011
- ↑ a b Sovers OJ, Kern CW, Pitzer RM, Karplus M. Bondfunction analysis of rotational barriers: ethane. J Chem Phys 1968, 49:2592–2599.
- ↑ Bader RFW, Cheeseman JR, Laidig KE, Wiberg KB, Breneman C. Origin of rotation and inversion barriers. J Am Chem Soc 1990, 112:6530-6536
- ↑ Havenith, R. W. A.; Van Lenthe, J. H. Delocalization in Valence Bond—Hyperconjugation. DOI 10.1002/qua.22018.
- ↑ a b Lowe JP. Simple molecular orbital explanation for the barriers to internal rotation in ethane and other molecules. J Am Chem Soc, 1970, 92: 3799-3800.
- ↑ Brunck TK, Weinhold F. Quantum mechanical studies on the origin of barriers to internal rotation about single bonds. J Am Chem Soc 1979, 101: 1700-1709
- ↑ Goodman L, Pophristic, V, Weinhold F. Origin of methyl internal rotation barriers. Acc Chem Res 1999, 32: 983-993.
- ↑ Pophristic V, Goodman L. Hyperconjugation not steric repulsion leads to the staggered structure of ethane. Nature, 2001, 11:7091-7101.
- ↑ Mo Y, Gao J Theoretical Analysis of the Rotational Barrier of Ethane. Acc. Chem. Res. 2007, 40(2), 113-119
- ↑ Weinhold, F. in The Encyclopedia of Computational Chemistry (ed. Schleyer, P. v. R.) 1792±1811 (John Wiley & Sons, Chichester, 1998).
- ↑ Rogers DW, Matsunaga N, Zavitsas AA, McLafferty FJ, Liebman JF. The conjugation stabilization of 1,3- butadiyne is zero. Org Lett 2003, 5:2373–2375.
- ↑ Rogers DW, Matsunaga N, McLafferty FJ, Zavitsas AA, Liebman JF. On the lack of conjugation stabilization in polyynes (polyacetylenes). J Org Chem 2004, 69:7143–7147.
- ↑ Kistiakowsky GG, Ruhoff JR, Smith HA, Vaughan WE. Heats of organic reactions. IV. Hydrogenation of some dienes and of benzene. J Am Chem Soc 1936, 58:146–153.
- ↑ Jarowski PD, Wodrich MD, Wannere CS, Schleyer PVR, Houk KN. How large is the conjugative stabilization of diynes? J Am Chem Soc 2004, 126:15036–15037.
- ↑ Cappel D, Tullmann ST, Krapp A, Frenking G. Direct estimate of the conjugative and hyperconjugative stabilization in diynes, dienes, and related compounds. Angew Chem Int Ed Engl 2005, 44:3617– 3620.
- ↑ Lide DR Jr, Mann DE Microwave spectra of molecules exhibiting internal rotation. I.propylene. J Chem Phys 1957, 27:868–873
- ↑ Lin BL, Xie Z, Liu R, Liu L. Guo QX. Importance of σ-type hyperconjugation to the eclipsed structure of propylene. JMol Struct: Theochem 2003, 633:15–19.
- ↑ Jalbouta AF, Basso EA, Pontesc RM, Dasa D. Hyperconjugative interactions in vinylic systems: the problem of the barrier to methyl rotation in acetone.JMol Struct: Theochem 2004, 677:167–171.
- ↑ Alabugin IV, Zeidan TA. Stereoelectronic effects and general trends in hyperconjugative acceptor ability of σ bonds. J Am Chem Soc 2002, 124:3175–3185.
- ↑ Apeilog Y, Schleyer PVR, Pople JA. Molecular orbital theory of the electronic structure of molecules. 35. β -Substituent effects on the stabilities of ethyl and vinyl cations. Comparison with isoelectronic methyl boranes. The relative importance of hyperconjugative and inductive effects. J AmChemSoc 1977, 99:5901–5909.
- ↑ a b Huang, Y., Zhong, A., Origin of anomeric effect: A density functional steric analysis, The Journal of Chemical Physics, 2011, 134, 084103.
- ↑ a b c Yirong Mo, Nature Chemistry, 2(8), 666-671, 2010.
- ↑ Ying Huang, Ai-Guo Zhong, Qinsong Yang, Shubin Liu. The Journal of Chemical Physics, 134, 084103, 2011.
- ↑ Tao Liu, Fang Yuan, Bing-Chao Li, Zhang-Yu Yu, Journal of Molecular Structure: THEOCHEM, 951, 82-88, 2010.
- ↑ Edward, J.T., Chem. Ind. (London), 1955, 1107.
- ↑ Anderson, C.B., Sepp, D.T., J. Org. Chem., 1967, 32, p.607.
- ↑ Wolfe, S. et al., J Chem Soc. B, 1971, 136.
- ↑ Lemieux, R.U., Pavia, A.A., Martin, J.C., Wantanabe, K.A., Solvation Effects on Conformational Equilibria. Studies Related to the Conformational Properties of 2-Methoxytetrahydropyran and Related Methyl Glycopyranocides., Can. J. Chem., 1969, 47, p.4427-4439.
- ↑ Juaristi, E., Cuevas, G., Recent Studies of the Anomeric Effect, Tetrahedron, 1992,48, p.5019-5087.
- ↑ David, S. et al., Superjacent orbital control, J Am Chem Soc, 1973, 95, p.3806-3807.
- ↑ Reed, A.E., Schleyer, P.V.R., The anomeric effect with central atoms other than carbon. 2. Strong interactions between nonbonded substituents in mono- and polyfluorinated first- and second-row amines, Inorganic Chem, 1988, 27, p.3969-3987
- ↑ Cramer, J.C., Hyperconjugation as it affects conformational analysis, J Molecular Struct (Theochem), 1996, 370, p.135-146.
- ↑ Anderson, J.E., Cai, J., NMR study of stereoelectronic anomeric and homoanomeric effects on the axial and equatorial C-H bonds in 1,3-diazacyclohexanes and 1,5-diazabicyclo[3.2.1]octanes, J. Chem. Soc. Perkin Trans, 1997, 12, p.2633-2637
- ↑ Fuson, R.C., Zirkle, C.L., Ring enlargement by rearrangement of the 1,2-aminochloroalkyl group; rearrangement of 1-ethyl-2-(chloromethyl)pyrrolidine to 1-ethyl-3-chloropiperidine, J. Am. Chem. Soc., 1948, 70, p.2760-2762.
- ↑ Parr, R.G., Yang, W., Density Functional Theory for Atoms and Molecules (Oxford University Press, New York, 1989).
- ↑ Liu, T. et al., J. Molecular Struc (THEOCHEM), 2010, 951, p.82-88
- ↑ Guerra CF, Baerends EJ, Bickelhaupt FM. Orbital interactions and charge redistribution in weak hydrogen bonds: Watson-Crick GC mimic involving C H proton donors and F proton acceptor groups. Int J Quantum Chem 2006, 106:2428–2443.
- ↑ Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 1988, 88:899–926.
- ↑ a b Alabugin IV,Manoharan M. Radical-anionic cyclizations of enediynes: remarkable substituent effects in cyclorearomatization reactions. J Am Chem Soc 2003, 125:4495–4509.
- ↑ Wolfe S. Gauche effect. Stereochemical consequences of adjacent electron pairs and polar bonds. Acc Chem Res 1972, 5:102–111
- ↑ Goodman L,Gu H, Pophristic V. Gauche effect in 1,2-difluoroethane. Hyperconjugation, bent bonds, steric repulsion. J Phys Chem A 2005, 109:1223–1229.
- ↑ Buissonneaud DY, van Mourik T, O’Hagan D. A DFT study on the origin of the fluorine gauche effect in substituted fluoroethanes. Tetrahedron 2010, 66:2196–2202.
- ↑ Schuler M, O’hagan D, Slawin AMZ. The vicinal F-C-C-F moiety as a tool for influencing peptide conformation. Chem Commun (Camb) 2005,34:4324–4326.
- ↑ Tavasli M, O’hagan D, Pearson C, Petty MC. The fluorine gauche effect. Langmuir isotherms report the relative conformational stability of (+/−)-erythro- and (+/−)-threo-9,10-difluorostearic acids. Chem Commun (Camb) 2002, 11:1226–1227.
- ↑ Souza FR, Freitas MP, Rittner R. On the stereoelectronic effects governing the rotational isomerism of 1,2-dihaloethanes. J Mol Struct: Theochem 2008, 863:137–140.
- ↑ Pitzer KS, Hollenberg JL. Cis- and trans- dichloroethylenes. The infrared spectra from 130 to 400 cm−1 and the thermodynamic properties. J Am Chem Soc 1954, 76:1493–1496.
- ↑ Kanakaraju K, Senthilkumar K, Kolandaivel P. Origin of the cis effect-nonbonded intramolecular interactions: quantum chemical studies on 1,2-dihaloethylene molecules. J Mol Struct: Theochem 2002, 589–590:95–102.
- ↑ Yamamoto T, Kaneno D, Tomoda S. The origin of cis effect in 1,2-dihaloethenes: the quantitative comparison of electron delocalizations and steric exchange repulsions. Bull Chem Soc Japan 2008, 81:1415–1422.








